


Blog by Martha Herbert
July, 2014
A summary version of this blog was posted by Autism Speaks on July 28, 2014
at http://www.autismspeaks.org/blog/2014/07/28/more-prenatal-genetics-search-autism%E2%80%99s-origins
AUTISM AND BRAIN TISSUE: POTENTIAL AND CHALLENGES
We will all benefit from better understanding of what is happening in the brain to drive the features of autism, and how the brain came to be that way. Studying tissue specimens from the brains of people diagnosed with ASDs who have died and whose families have donated their brains to research is one of the most poignant and precious ways to do this. The huge power of this approach is the lens it gives us into the microscopic level –the cells of the brain and the parts of those cells – and how they may differ from the same components in the brains of people without ASDs. Other than the very rare opportunity to obtain brain tissue from surgeries, there is no way to get this level of information from living human beings.
At the same time, these specimens are from people who are no longer alive. This means that we cannot observe their behavior or check out hunches about how these cellular changes might have contributed to the particular ways their autism worked. And we cannot know for sure exactly when in the course of their lives these changes may have developed.
Another challenge is that people with ASDs are not diagnosed until well after birth – and we have no biological tests to predict this diagnosis, so that it is essentially impossible to get samples of brain tissue from people who “are autistic” if they haven’t been behaviorally diagnosed. Moreover, people with autism don’t often die young, making it rare for young children with autism to not only to die but also to have their brains donated for research. As a result, studies of brain tissue have most often been performed on samples from older individuals – late childhood to well into the adult lifespan. Using such data to make claims about early origins of autism is done by “reading back in time;” but such claims are only inference – not fact.
CONTRIBUTIONS OF A RECENT PAPER
The recent New England Journal of Medicine paper by Stoner and colleagues, “Patches of disorganization in the neocortex of children with autism.” N Engl J Med 370(13): 1209-1219, made several important contributions to studies of postmortem tissue samples from the brains of individuals with ASDs. First, the average age was much younger than the average age in previous studies. Second, many important and technologically sophisticated measures were made. Finally, as a result of this work, a broadly distributed pattern of brain tissue abnormalities was identified that had not been described on this scale before – namely, patches of disorganization in many areas of the neocortex (the outer layer of gray matter in the brain), though not the same from one person to the next.
CRITICAL QUESTIONS
The new observation of disorganized patches by itself is very important. It poses an important challenge: how do we figure out what it means? To do that, we need to understand the data, and also to examine the interpretations and consider alternative possibilities.
- To UNDERSTAND the data, we need to ask: WHAT the changes in these patches are as compared to normal neocortex, WHERE these changes occurred around the brain, WHEN the causative process or processes occurred, HOW they were caused, and what IMPACT they have on brain FUNCTION.
- To EVALUATE THE INTERPRETATIONS, we need to see if the data presented could support interpretations other than those offered by the authors. This requires examining underlying assumptions and considering alternative frameworks that might make different – and perhaps better – sense of the data. This step is critical, because HOW these questions are answered has huge implications for how we prioritize our efforts in autism research and treatment.
WHAT WERE THE CHANGES IN THE WAY THE CELLS WERE ORGANIZED?
The neocortex of the brain (which is made up mainly of neurons) for the most part has six distinct cellular layers. In these patches they found various kinds of disruption of this layering. They found disturbances both in the structure of the tissue and in markers for several important proteins. These patches were found in tissue from 10 of 11 children affected by autism and 1 of 11 unaffected children.
They observed that while the type of disorganization tended to be similar among patches from the same person’s brain, there was less consistency when they compared patches between different people. Yet even when comparing different patches from the same person, no two patches were identical in their presentation. So this was not a uniform, cookie-cutter type change at all.
A reduction in the markers for excitatory neurons seemed to be the best indicator of the presence of a patch, but several other types of markers sometimes also showed abnormalities.
WHERE WERE THE CHANGES LOCATED AROUND THE BRAIN?
The investigators were able to obtain tissue samples from four regions of the brain. Though this is a small fraction of the total surface are of the brain, small samples (both in amount of tissue and number of individuals studied) are standard for brain tissue research since brain tissue is so precious and rare. They found many patches in the dorsolateral prefrontal cortex in 10 of 11 cases with ASD, and also in the 2 of 2 ASD cases in the posterior superior temporal cortex. They did not find abnormal patterns of marker expression in the occipital cortex (where they had 3 case samples). One individual without autism (out of 11) had some patches in the prefrontal cortex.
Although several neuropathologists who commented on this paper on the SFARI website expressed concerns that these patches might be artifacts resulting from tissue preparation and storage processes, theystill thought it was notable that there were more patches in the samples from people with autism than from those with neurotypical development.
My MRI brain imaging team felt sympathetic to the findings themselves because some of our analyses suggest similar-sized randomly distributed patchy areas of thinning, as well as thickening, as illustrated in the figure. Our findings cannot be directly comparable to what the Stoner et al. study reports, because the Stoner et al. study focused more on genetic marker changes than on physical thinning, whereas MRI can measure the latter but not the former.
On the other hand, MRI allows us to visualize the neocortex of the entire living brain, which is not possible to do with post-mortem brain tissue samples. We see patches all over the brain, which makes us suspect that the Stoner et al. prediction that the abnormalities will be “detectable in the prefrontal and temporal cortex” and imply—based on only 3 samples– that they won’t be found occipital lobe of the brain. While this prediction fits with their theory that prefrontal and temporal cortex are especially central to autism, we doubt that it will be borne out.
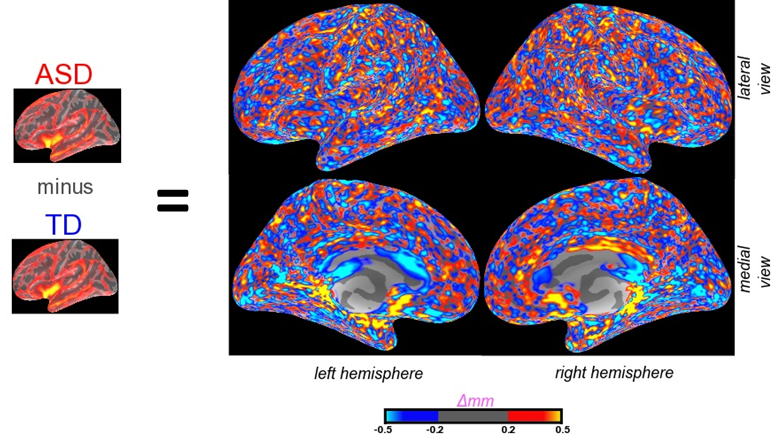
| Figure legend: To generate the figures and measures of patches using MRI, we subtracted the average cortical thickness at each point (as determined by Freesurfer imaging analysis software) of a typically developed group of subjects (TD) from the average cortical thickness at each analogous point for a group of subjects with ASD. These patches can also be visualized and measured at a lesser density in scans of individual brains. |
WHEN DID THESE CHANGES OCCUR?
The layers of the neocortex develop through a process called “lamination.” This process takes place early in fetal development. This process, which occurs through a very interesting “inside out” pattern, has been very carefully studied. Clearly one possible way that these patches of disorganization could have been created is by failure of the lamination process to proceed properly in the brain in the first place. The investigators focused on this possibility, particularly since they had previously identified genetic changes that can be related to these processes.
However, the alternative possibility, that the brain’s layers developed normally but then something happened to them, was not systematically discussed or investigated. It is certainly possible that some of these patches of missing genetic markers could emerge during the initial development of cortical layering. But it is also possible that the tissue involuted – was damaged or or degraded – after its initial development. Until this possibility is systematically investigated, it cannot be dismissed, and the evidence presented in this study simply does not support either definitively declaring that the patches developed only in pregnancy, only through genetic causes, or excluding or ignoring the possibility that some or many of them could have originated later and/or in other ways.
HOW MIGHT THESE CHANGES HAVE OCCURRED? NARROW FIELD OF VIEW OF THE INVESTIGATORS
The investigators clearly stated in their paper that they had not identified the specific mechanisms leading to the presence of these patches of disorganization. But their discussion of possible mechanisms was confined to an exclusive focus on alterations of processes occurring as the brain’s layers developed in the first place. This assumption narrowed many of their choices in how they designed their experiments around missing markers. The genetic markers they chose were ones that could identify changes in cortical layers and in the types of cells present. They also looked at genes associated with autism from prior research. Starting with 63 genes and then narrowing the list down (by screening them in two boy cases) to 25 genes that were present most consistently and robustly, they mainly selected genes that have easy-to-observe expression patterns that are also markers of cell types (types of neurons, glial cells, etc.), and pertinent particularly to processes early in development.
It is important to remember that while data from measurements can be “objective,” the choice of what to measure is not objective, but reflects underlying assumptions that need to be (but in this case were not) tested against alternative assumptions.
HOW ELSE MIGHT THESE PATCHES BE FORMED OTHER THAN A GENETIC ALTERATION OF INITIAL LAYERING?
Development can be altered not only by genetics but by many non-genetic factors that are now document as increasing risk for autism. These include physiological problems such as various maternal health problems (infections, immune and metabolic disturbances such as maternal diabetes, obesity and high blood pressure) 1,2 and environmental exposures such as pesticides and other toxicants (which may build up in the placenta and get passed on to the developing fetus) 3-6 7-9 as well as maternal stress. 10,11 Insufficient supply or imbalances of nutrients such as vitamin D, antioxidants, minerals such as magnesium and zinc or essential fatty acids may contribute as well. 12,13 Braun, 2014 #4021}14 though studies are limited. Other environmental exposures (such as electromagnetic fields or radiofrequency radiation are known to degrade the health of cells and metabolism before conception, prenatally and postnatally in ways that look like what has been documented in autism (e.g., mitochondrial damage, lipid peroxidation) but are only starting to be studied in cohorts with autism. 15-17
Certain genetic changes could lead to greater vulnerability to these problems,18 but to actually develop problems you need the environmental contribution of not getting enough of something your system needs, or getting too much of something your system can’t handle well.
Such non-genetic factors environmental and physiological factors, individually or in combination, could be contributors to changes in utero that might impact the brain, by leading to vulnerabilities in brain cells, membranes and tissue – and such weaknesses would make the baby’s brain more easily injured during pregnancy or infancy. 19 Causation by these types of contributors might, if anything, be more consistent with the structure and distribution of these regions of disorganization – which are different between individuals, not even entirely consistent between regions in the same individuals, and distributed fairly randomly and differently from person to person.
How might such physiological, structural and environmental risk factors influence the layers of the neocortex? Although this hasn’t been studied directly in ASDs directly in relation to early brain development, we know a lot about what these factors can do under other circumstances. They can alter or interfere with blood flow (through compromise of small vessels and/or increase in blood viscosity—stickiness—from metabolic challenges), and they can change cell function due to reactions to infection, toxic exposures or exposure to abnormal metabolic or immune reactions and/or brain chemistry. The disorganization of the layers could also potentially be caused by inflammation which, perhaps because of the swelling it involves, may introduce shear forces that tend to pull apart the tissue, affect the folding patterns of the cortex, and cause nicks and ruptures in tissue. Any of these, or a combination of these problems, could potentially contribute to disrupting brain tissue health throughout the lifespan.
The way these processes may disrupt tissue is not through primary genetic mechanisms in tissue development but through degradation. Stoner et al. have identified gene changes and missing genes, but they seem to assume that this proves that the patchy changes are caused by genes. Actually, gene findings don’t prove at all that genetic processes are the primary cause of gene changes. We must remember that genetic damage and gene expression changes can occur on account of secondary processes too. Both can result from environmental and physiological stressors. The randomness of where these patches of missing gene expressions show up, and the variation in how they are structured – these are if anything more consistent with physiological or environmental insults than with genetic programming.
It is interesting that the authors say they set out to find a genetic cause for the “brain disturbance” in autism which they assumed (though this has never been proven) to be comparable to various serious and rare disturbances of brain development such as schizencephaly. But even in those conditions, substantial scientific literature supports contributions besides genetics, including trauma, disturbance of blood flow, early stroke and infection. 20,21 This is another example of how their assumptions have narrowed their field of view.
WHAT IMPACT MIGHT THESE TISSUE CHANGES HAVE ON BRAIN FUNCTION?
We need to look at functional impact at both the cellular and the information systems levels of the brain.
At the cellular level, what can we make of the reduction in markers for excitatory neurons? There are possible genetic contributors, but we also need to take account of the fact that some of the physiological changes (such as oxidative stress, mitochondrial dysfunction and immune abnormalities) – that that are well documented in people with autism, and also sometimes – perhaps often – found in their mothers and fathers – appear to associated with diseases characterized by increase excitatory activity in the brain. 22-25 It is also well known that such excessive excitatory activity can be caused by environmental factors including the above-listed risk factors for autism. 26 When brain cells are overly excited for too long, the situation becomes “excitotoxic” – the cells can get sick and overwhelmed, and even die from “burnout.” 27-29 Such issues are well known to arise in neurodegenerative diseases in older adults. 30-32 This kind of possible scenario for (de)generation of these patches of disorganized neocortex was not considered in the Stoner et al. paper. For the sake of affected individuals, those at risk and of science, I think it should be seriously investigated.
At the information systems level, the authors speculate in their discussion that the variable location of these disorganized patches of neocortex could contribute to heterogeneity in ASDs, by disrupting different functional systems from one person to the next. While that is possible, this makes the assumption that the changes in brain molecular and cellular structure in these patches drive brain function in a one-way direction.
If, from the above alternative scenario, cellular dysfunction is what triggered these patches to form in the first place, it could also work the other way around. In this scenario, this cellular dysfunction has no reason to “go away” after causing harm in the cortex. Instead, it could stick around and continue to disturb brain function, irritating synapses and altering brain networks, on an ongoing basis into infancy and childhood. The structural disruptions could play a role too, but in a secondary way.
This possibility–that the “autism” is generated in an ongoing, moment-to-moment fashion by cells that have taken hits from environmental risk factors–has massive implications for autism research and treatment that need to be not ignored, but explored–indeed pursued actively. This means that we need to start thinking about the impact of environmental influences as not simply “flipping a switch” and causing the “autism” that then runs on auto-pilot for life, but instead as generating the autism at every moment by changing how many components of brain tissue (including not just neurons but also glial cells, vascular supply and extracellular matrix) contribute to synaptic activity and network coordination. This also means that by improving the health of these brain components, we might be able to improve brain function. This has major implications that go beyond genetics and medicine to health and environment.
CONCLUSION AND RECOMMENDATIONS
It is very important to keep alive the question of how and when autism arises, and to consider environmental and physiological factors and not just the genetic contributors on which Stoner et al., and so many others. The idea that environmental and physiological influences are subsidiary or minor additions to the overwhelming role of genes is a belief, not a fact. We need to keep a broader perspective alive not only to explain what is driving the increasing numbers of people with autism, but also because there are things we already know about how to help reduce many of these risk factors, and doing these things could reduce the numbers and severity of ASDs substantially. Many of the environmental exposures and the physiological problems that environmental exposures trigger (such as inflammation, metabolic syndrome, mitochondrial dysfunction, nutrient deficiencies and oxidative stress) are risks that can be greatly reduced by public health and lifestyle interventions that we have enough knowledge to implement right now.
Neurodevelopmental processes should be a question, not an answer. We should not simply assume that if it is neurodevelopmental, it is purely genetic. Genetically disturbed neurodevelopment may be part of the picture in some people, but environmental, epigenetic and physiological contributors are highly likely to exist as well, and for many people they may be the larger part of the picture. Moreover these environmental contributors may contribute to the challenges of autism not only in creating the condition, but also in making it more complicated–and they may do create these complications not only in utero but also across the entire lifespan.
People with autism, their families, those suffering from chronic diseases with overlapping genetic and environmental contributors and physiological mechanisms, and society as a whole would benefit by a concerted attempt by the funders of autism research to take a broader, more comprehensive and more integrated approach to the science, the policy, and the education of the community and public, and to research autism and interpret scientific findings as if not just genetics but also environment and physiology really mattered.
CITATIONS
1. Suren P, Gunnes N, Roth C, et al. Parental Obesity and Risk of Autism Spectrum Disorder. Pediatrics 2014.
2. Xu G, Jing J, Bowers K, Liu B, Bao W. Maternal diabetes and the risk of autism spectrum disorders in the offspring: a systematic review and meta-analysis. J Autism Dev Disord 2014;44:766-75.
3. Volk HE, Lurmann F, Penfold B, Hertz-Picciotto I, McConnell R. Traffic-related air pollution, particulate matter, and autism. JAMA Psychiatry 2013;70:71-7.
4. von Ehrenstein OS, Aralis H, Cockburn M, Ritz B. In Utero Exposure to Toxic Air Pollutants and Risk of Childhood Autism. Epidemiology 2014.
5. Rossignol DA, Genuis SJ, Frye RE. Environmental toxicants and autism spectrum disorders: a systematic review. Transl Psychiatry 2014;4:e360.
6. Shelton JF, Geraghty EM, Tancredi DJ, et al. Neurodevelopmental Disorders and Prenatal Residential Proximity to Agricultural Pesticides: The CHARGE Study. Environ Health Perspect 2014.
7. Shelton JF, Hertz-Picciotto I, Pessah IN. Tipping the balance of autism risk: potential mechanisms linking pesticides and autism. Environ Health Perspect 2012;120:944-51.
8. D’Amelio M, Ricci I, Sacco R, et al. Paraoxonase gene variants are associated with autism in North America, but not in Italy: possible regional specificity in gene-environment interactions. Mol Psychiatry 2005;10:1006-16.
9. Roberts EM, English PB, Grether JK, Windham GC, Somberg L, Wolff C. Maternal residence near agricultural pesticide applications and autism spectrum disorders among children in the California Central Valley. Environ Health Perspect 2007 Oct;115(10):1482-9 2007.
10. May-Benson TA, Koomar JA, Teasdale A. Incidence of pre-, peri-, and post-natal birth and developmental problems of children with sensory processing disorder and children with autism spectrum disorder. Front Integr Neurosci 2009;3:31.
11. Beversdorf DQ, Manning SE, Hillier A, et al. Timing of prenatal stressors and autism. J Autism Dev Disord 2005;35:471-8.
12. Lyall K, Schmidt RJ, Hertz-Picciotto I. Maternal lifestyle and environmental risk factors for autism spectrum disorders. Int J Epidemiol 2014;43:443-64.
13. Schmidt RJ, Hansen RL, Hartiala J, et al. Prenatal Vitamins, One-carbon Metabolism Gene Variants, and Risk for Autism. Epidemiology 2011;22:476-85.
14. Jones KL, Will MJ, Hecht PM, Parker CL, Beversdorf DQ. Maternal diet rich in omega-6 polyunsaturated fatty acids during gestation and lactation produces autistic-like sociability deficits in adult offspring. Behav Brain Res 2013;238:193-9.
15. Herbert MR, Sage C. Autism and EMF? Plausibility of a pathophysiological link – Part I. Pathophysiology 2013;20:191-209.
16. Herbert MR, Sage C. Autism and EMF? Plausibility of a pathophysiological link – Part II. Pathophysiology 2013;20:211-34.
17. Alsaeed I, Al-Somali F, Sakhnini L, et al. Autism-relevant social abnormalities in mice exposed perinatally to extremely low frequency electromagnetic fields. Int J Dev Neurosci 2014;37C:58-64.
18. Herbert MR. Contributions of the environment and environmentally vulnerable physiology to autism spectrum disorders. Curr Opin Neurol 2010;23:103-10.
19. Herbert MR. Why aren’t we there yet? Valuable but incomplete measures of brain changes in babies with autism. Autism Why and How www.autismWHYandHOW.org 2012.
20. Pascual-Castroviejo I, Pascual-Pascual SI, Velazquez-Fragua R, Viano J, Quinones D. Schizencephaly: a study of 16 patients. Neurologia 2012;27:491-9.
21. Sener RN. Schizencephaly and congenital cytomegalovirus infection. Journal of neuroradiology Journal de neuroradiologie 1998;25:151-2.
22. de la Monte SM, Tong M. Brain metabolic dysfunction at the core of Alzheimer’s disease. Biochemical pharmacology 2014;88:548-59.
23. Blaylock RL, Maroon J. Immunoexcitotoxicity as a central mechanism in chronic traumatic encephalopathy-A unifying hypothesis. Surg Neurol Int 2011;2:107.
24. Yates KF, Sweat V, Yau PL, Turchiano MM, Convit A. Impact of metabolic syndrome on cognition and brain: a selected review of the literature. Arteriosclerosis, thrombosis, and vascular biology 2012;32:2060-7.
25. Yang Y, Song W. Molecular links between Alzheimer’s disease and diabetes mellitus. Neuroscience 2013;250:140-50.
26. Lubrano C, Genovesi G, Specchia P, et al. Obesity and metabolic comorbidities: environmental diseases? Oxid Med Cell Longev 2013;2013:640673.
27. Essa MM, Braidy N, Vijayan KR, Subash S, Guillemin GJ. Excitotoxicity in the pathogenesis of autism. Neurotox Res 2013;23:393-400.
28. Ghanizadeh A. Targeting Mitochondria by Olesoxime or Complement 1q Binding Protein as a Novel Management for Autism: A Hypothesis. Mol Syndromol 2011;2:50-2.
29. Skaper SD, Floreani M, Ceccon M, Facci L, Giusti P. Excitotoxicity, oxidative stress, and the neuroprotective potential of melatonin. Ann N Y Acad Sci 1999;890:107-18.
30. Beal MF. Role of excitotoxicity in human neurological disease. Curr Opin Neurobiol 1992;2:657-62.
31. von Bernhardi R, Eugenin J. Alzheimer’s disease: redox dysregulation as a common denominator for diverse pathogenic mechanisms. Antioxid Redox Signal 2012;16:974-1031.
32. Dawson R, Jr., Beal MF, Bondy SC, Di Monte DA, Isom GE. Excitotoxins, aging, and environmental neurotoxins: implications for understanding human neurodegenerative diseases. Toxicol Appl Pharmacol 1995;134:1-17.
